
The projects of our team* focus on triple-negative breast cancer (TNBC) for which the discovery of treatments is a societal need. Our integrated and multidisciplinary research aims (i) at the fundamental level, to determine how the microenvironment of TNBC is affected by proteases, matricellular proteins and bioactive fragments released by the N-TAILS proteomics; (ii) at the translational level, to modulate the tumor/stroma crosstalk by targeting identified proteins and bioactive fragments with proprietary antibodies and antibody-drug conjugates (ADC); and (iii) at the pre-clinical level, to select TNBC subgroups for targeted and/or combined treatment with antibody. Our primary objective is to translate our fundamental discoveries into clinical applications to treat certain subtypes of TNBC for personalized medicine.
Axis 1 : Extracellular matrix (ECM) protumor reprogramming through the tumor-stroma crosstalk in triple-negative breast cancers (TNBC) via ECM protein dysregulation/cleavage (leader, E Liaudet, DR2 INSERM).
To dissect the tumor-stroma crosstalk modulated by proteolysis, we are characterizing the cathepsin D substrate repertoire by OMIC deep analysis in the TNBC microenvironment. Using N-TAILS degradomics, we discovered ~150 extracellular secreted proteins, including proteases, matricellular proteins, receptors and other components (and their related peptides), potentially implicated in the TNBC tumor microenvironment organization. These large datasets are analyzed by bioinformatic approaches to predict candidate substrates and to assess their coordination at the molecular level by biochemical approaches in order to identify new targets for antibody-based therapy. Using N-TAILS proteomic approach, we demonstrated that the matricellular protein SPARC is cleaved in its carboxy-terminal part by cath-D, releasing a 9-kDa SPARC fragment with exacerbated oncogenic activity compared with native SPARC in TNBC [Alcaraz et al, Theranostics, 2021]
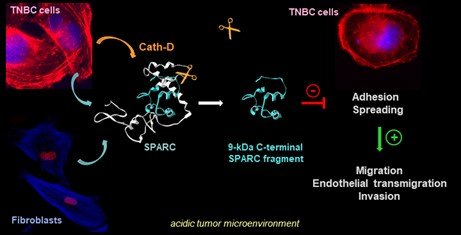
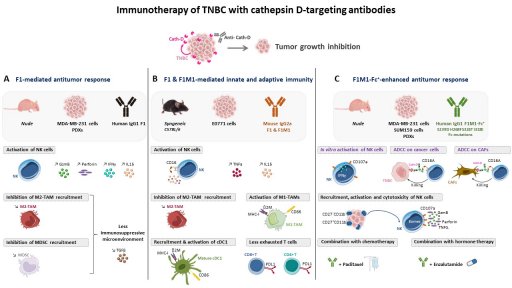
Axis 2 : Innovative antibody-based therapies shaping the tumor microenvironment by targeting both cancer and stromal cells, for TNBC novel therapy (porteurs, T Chardès, DR1 Cnrs, V Laurent-Matha, MCU, E Liaudet).
TNBC treatment is mainly restricted to chemotherapy. Hence, tumor-specific molecular targets and/or alternative therapeutic strategies for TNBC are urgently needed. Immunotherapy and antibody-drug conjugates (ADC) are emerging as exciting treatment options. We select potential interesting targets from the tumor microenvironment according to our N-TAILS degradomics to develop innovative human antibodies and ADC for TNBC novel therapy. Our recent studies have revealed the anti-tumor and immunomodulatory activity of our proprietary human anti-cathepsin D antibodies [Desroys du Roure*, David*, J. Immunother. Cancer, Commentary, in press]
Axis 3 : Selection of patients with TNBC who could benefit from innovative antibody and ADC therapy (leaders: S Guiu,MD-PhD, breast cancer specialist, ICM, and P Roger, PU-PH, pathologist, CHU de Nîmes)
Due to TNBC heterogeneity, it is important to identify subgroups eligible for targeted therapies. A new TNBC classification, based on immunohistochemistry features, distinguishes four TNBC subgroups: luminal androgen receptor-positive (LAR), immunomodulatory (IM), basal-like immunosuppressive (BLIS), and mesenchymal (MES). With the team's medical doctors, we will analyze the expression of our targets, identified by N-TAILS and validated by functional targeting with proprietary antibodies, in this TNBC classification by immunohistochemistry using TNBC microarrays to identify TNBC subgroup(s) that could benefit from innovative antibody-based therapy and ADCs. Our previous preclinical study highlighted the LAR subgroup that may benefit from combination therapy with anti-androgens and anti-cathepsin D antibody [Mansouri*, Alcaraz* et al, Cancers, 2020].


Located in the Parc Euromédecine, the Institut de Recherche en Cancérologie de Montpellier (U1194) is located on the Val d'Aurelle Campus (ICM, Institut régional du Cancer Montpellier/ Val d'Aurelle).
U1194 Research Centre supported by Inserm, the University of Montpellier and the Institut du Cancer de Montpellier (ICM)




 Institut de Recherche en Cancérologie de
Institut de Recherche en Cancérologie de