
The p53 protein is the effector of an essential checkpoint regulating genome integrity, cell cycle progression, senescence and cell death that is frequently mutated in human cancers. Its tumor suppressive functions have been more recently extended to the control of cellular metabolism. We and others demonstrated that several other key components of this pathway (MDM2, MDM4, E4F1, ARF, BMI1) also display key metabolic activities both in a p53 dependent and independent manner. Our main objective is to further characterize the metabolic networks regulated by the p53 pathway and understand how their perturbation contributes to several pathophysiological conditions including metabolic disorders, aging, and cancer.
Axis 1 : Modelling of p53-associated metabolic networks (Project leader: L. Le Cam)
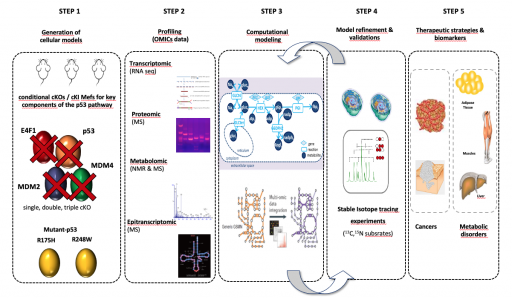
We are characterizing the metabolic networks regulated by the p53 pathway at an unprecedented resolution by profiling through a multi-omics approach (RNAseq, proteomics, metabolomics) primary cells derived from a unique collection of genetically engineered mouse models in which one, two or three key components of the p53 pathway are inactivated (Mdm2, Mdm4, E4f1, Trp53) or expressing different p53 mutants that are commonly found in human tumors (R175H et R248W). These large datasets are integrated into computational models that we use to identify new biomarkers or define new therapeutic strategies.
Axis 2 : Role of mutant p53 in the Li-Fraumeni syndrome (Project leader: L. Le Cam)
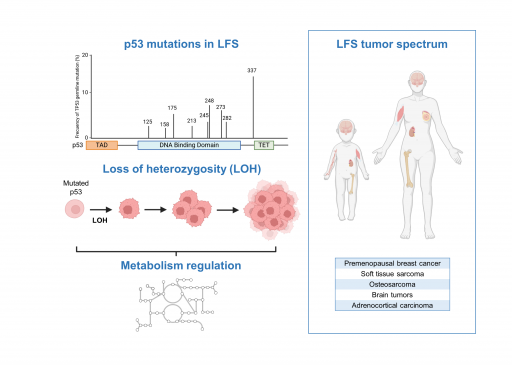
The Li-Fraumeni syndrome (LFS) is a rare but severe cancer predisposition syndrome linked to germline mutations of TP53. Through the characterization of LFS patient samples and of new animal models recapitulating some of the genetic alterations commonly found in the LFS, one of our main objectives is to further understand how mutated forms of p53 regulate metabolism during cancer progression.
Axis 3: Deregulation of p53-associated metabolic functions during aging (Project leader: PF. Roux)
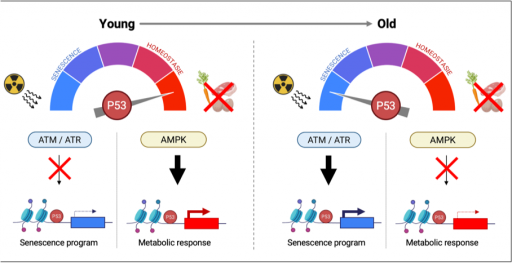
The p53 pathway plays a central role in regulating mechanism involved in aging, both at the cellular and organismal levels. p53 also participates in metabolic adaptations to physiological nutritional challenges such as those occurring during fasting. Using systems biology approaches, we explore how aging influences p53’s ability to regulate metabolism, thereby contributing to age-related diseases such as type II diabetes.
Axis 4: Role de metabolic heterogeneity in skin cancer development (Project leaders : M. Lacroix, E. Frouin, PE Stoebner).
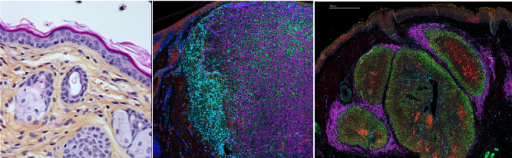
In this part of our team project, we are investigating the role of various metabolic networks regulated by key components of the p53 pathway in skin homeostasis and are evaluating how their deregulation contributes to the metabolic reprogramming of cancer cells in different types of skin cancers (melanoma, sebaceous carcinoma). We use patient samples as well as different mouse models (PDXs, GEMMs) that we characterize through multiplexed tissue imaging approaches (Hyperion and Xenium) to evaluate how metabolic changes contribute to tumor heterogeneity. One of our final objectives is to determine whether the metabolic zonation in those tumors define specific niches for populations of cancer cells contributing to metastatic dissemination and resistance to treatments.
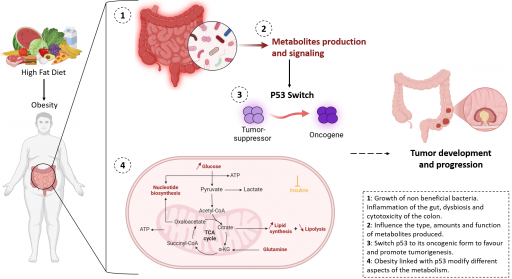
Axis 5: Metabolic exchanges between tumors and host (Project Leader: M. Lapierre)
During tumor initiation and progression, many metabolic changes influence the function of cancer cells to allow them to survive in unfavorable conditions (hypoxia, nutrient deprivation, etc…). Metabolite exchanges occur between cells of the tumor microenvironment but also between cancer cells and distant organs. Using original mouse models, tumoroids, and patient samples, this research axis addresses how metabolic changes occurring in the liver or in the adipose tissue, in particular those related to obesity, modulate the tumor ecosystem of p53-mutated colorectal cancers. This part of our project is based on several multiplexed imaging methods that we use to assess metabolism in situ.
Axis 6: Metabolic reprogramming of IDH-mutated tumors (Project Leader: L. Stuani)
In this axis, we aim at deciphering the mechanisms underlying the metabolic specificities, and their molecular consequences, of Isocitrate Dehydrogenase-mutated (IDHm) cancers, in particular gliomas (oligodendrogliomas and astrocytomas) and Acute Myeloid Leukemias (AML). Moreover, it was recently reported that the spectrum of TP53 mutations in IDHm is dominated by a single highly enriched hotspot mutation leading to the expression of the TP53R273C mutant. Using original models, we are trying to understand the rationale of this selective pressure leading to this combination of genetic alterations. Our final objectives are to develop new therapeutic strategies for this subclass of patients and to further understand the metabolic flexibility and plasticity of these cancer cells in different ecosystems.


Located in the Parc Euromédecine, the Institut de Recherche en Cancérologie de Montpellier (U1194) is located on the Val d'Aurelle Campus (ICM, Institut régional du Cancer Montpellier/ Val d'Aurelle).
U1194 Research Centre supported by Inserm, the University of Montpellier and the Institut du Cancer de Montpellier (ICM)




 Institut de Recherche en Cancérologie de
Institut de Recherche en Cancérologie de