
Le centre d’intérêt de l’équipe est l’inférence et l’exploitation de réseaux moléculaires afin d’extraire de l’information de jeux de données omiques complexes. Nous développons des méthodes et les appliquons au travers de collaborations avec des biologistes et médecins pour analyser des cohortes de tumeurs spécifiques de tailles limitées. Un accent particulier est mis sur le microenvironnement tumoral et ses réseaux cellulaires à partir de données bulk, en cellules uniques ou spatiales. Nous commençons à appliquer ces méthodes aux maladies autoimmunes. La science des réseaux et le machine learning (ML)/l’IA sont nos outils préférés pour ces travaux.
Nous développons aussi des méthodes relatives à la recherche de biomarqueurs. Dans domaine du cancer, nous travaillons principalement sur l’ADN circulant en utilisant le ML/IA. Nous employons aussi des méthodes d’analyse numérique et de modélisation bayésienne pour étudier la dynamique in vivo des protéines dans la maladie d’Alzheimer à la recherche de déviations.
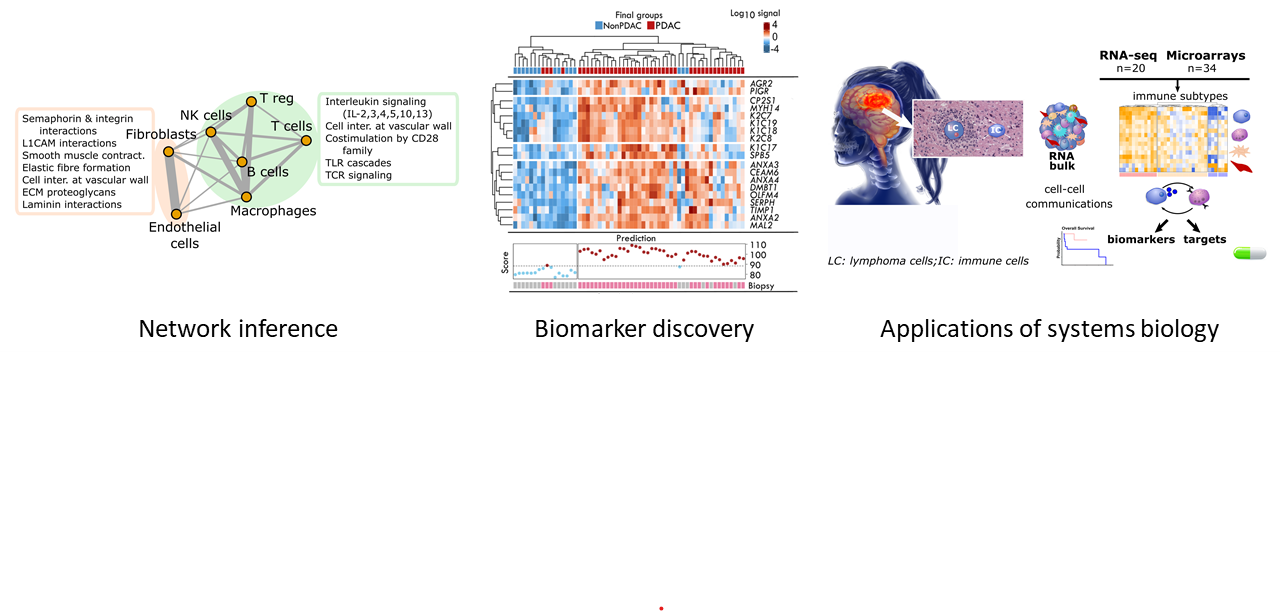
Axes de recherches :
Axe 1 : Inférence de réseaux moléculaires (Responsables : Ravel & Colinge)
L'analyse de données (multi-)omiques à grande échelle est souvent difficile. Contextualiser à l'aide de réseaux d'interactions facilite l'extraction d'informations fiables. Nous avons développé un framework original pour inférer des réseaux cellulaires et voies intracellulaires sous-jacentes à partir de données bulk, en cellules uniques ou spatiales (Cabello-Aguilar, NAR, 2020 ; Villemin, NAR, 2023). Nous continuons d'étendre ce framework avec des techniques d'IA notamment.
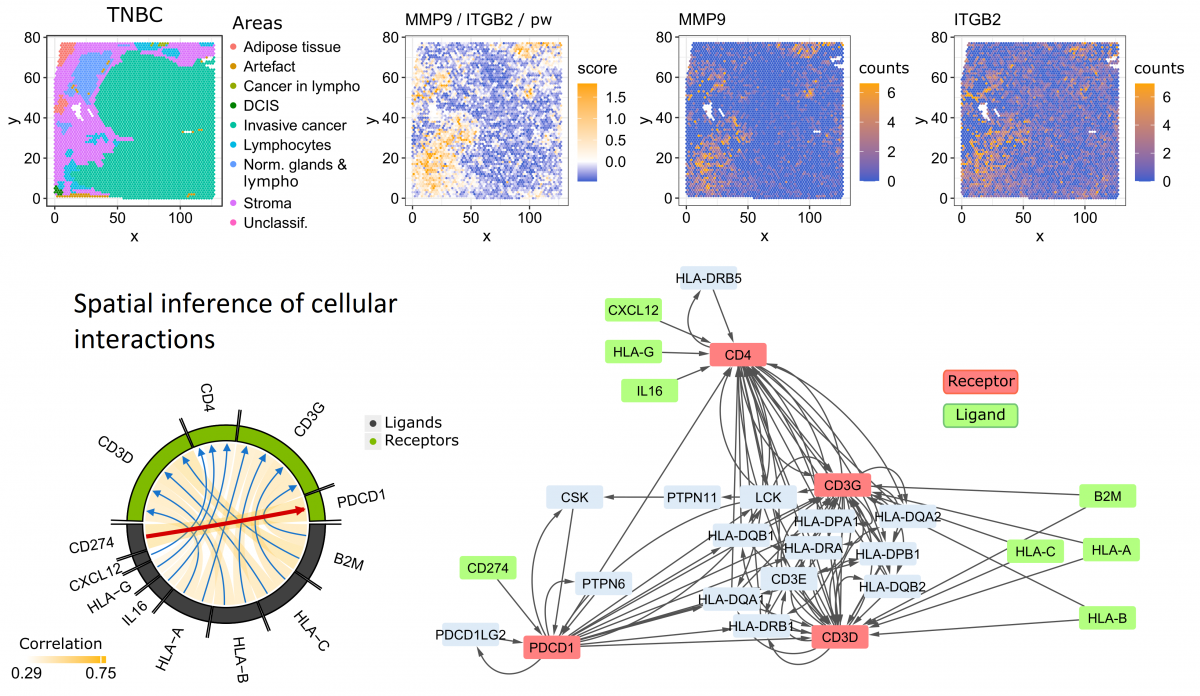
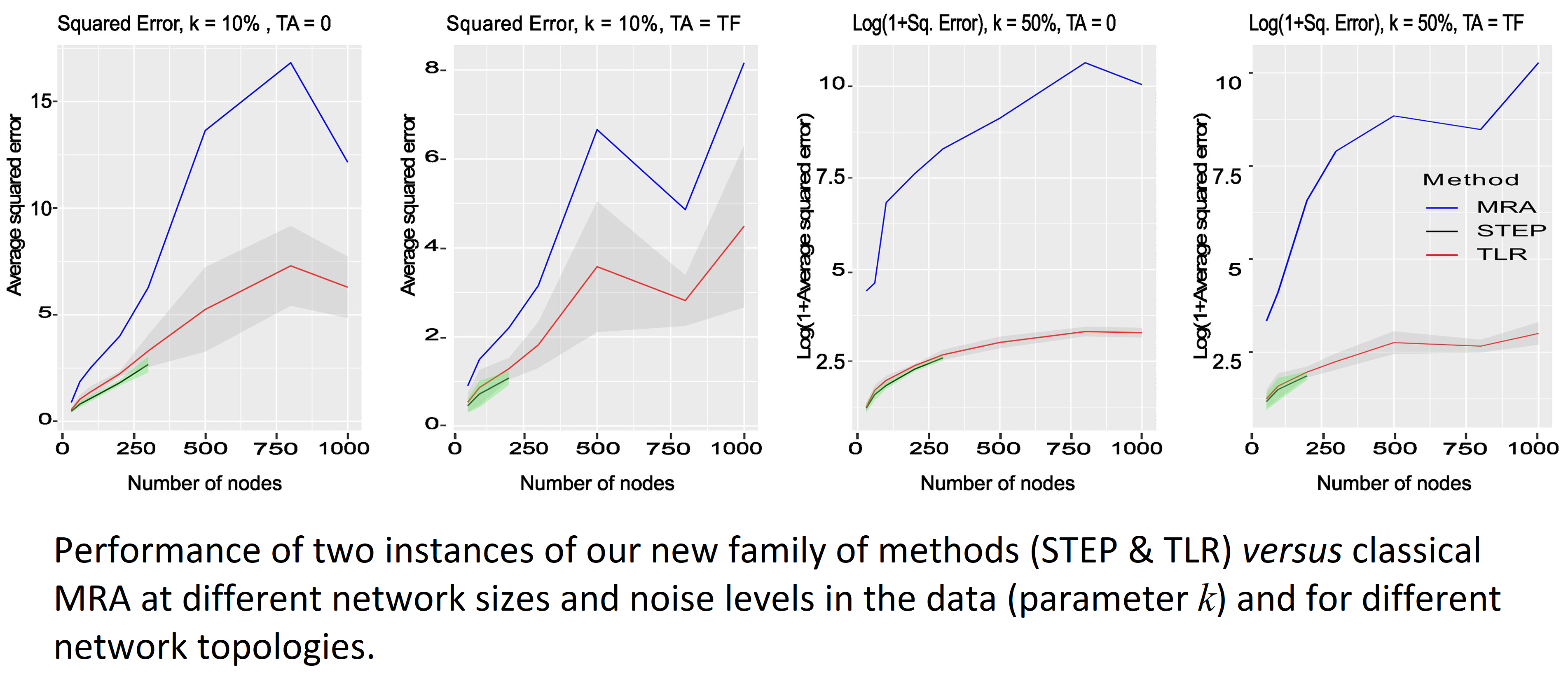
Nous avons également proposé une nouvelle famille de modèles statistiques pour déduire des interactions moléculaires à partir de données de perturbation (Borg, Bioinformatics 2023 & bioRxiv 2024). La généralisation de cette théorie est un sujet actif.
Axe 2 : Découverte de biomarqueurs par la modélisation mathématique (Responsable : Colinge)
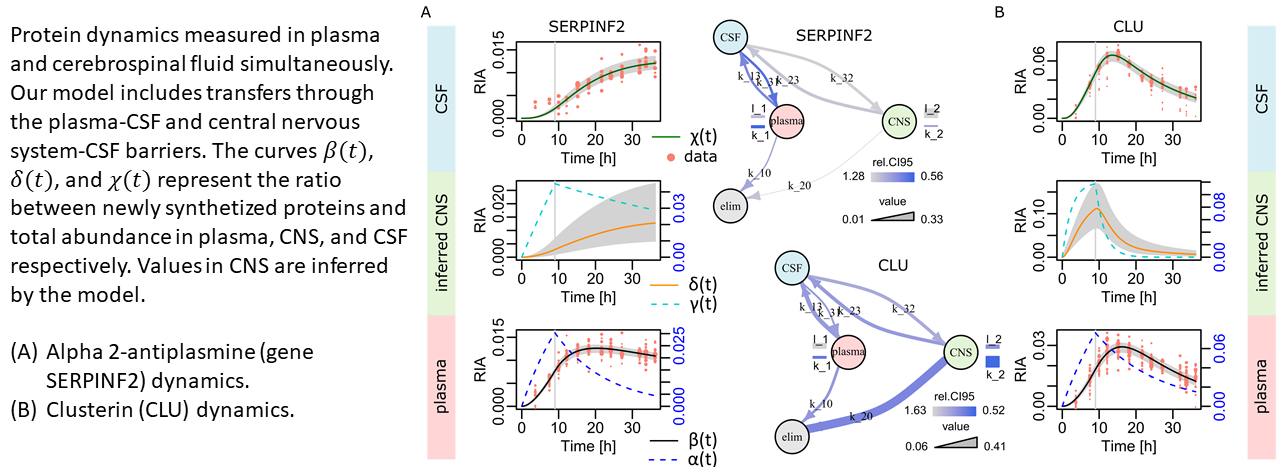
Appliquant des méthodes d'analyse numérique et de ML/AI, nous sommes engagés dans divers programmes de recherche de biomarqueurs. Un premier exemple concerne la recherche de dynamiques anormales des protéines dans la maladie d'Alzheimer (Lehmann, Anal Chem, 2019 ; Giroux, J Proteome Res, 2024 ; Lehmann, Bioinformatics, 2024).
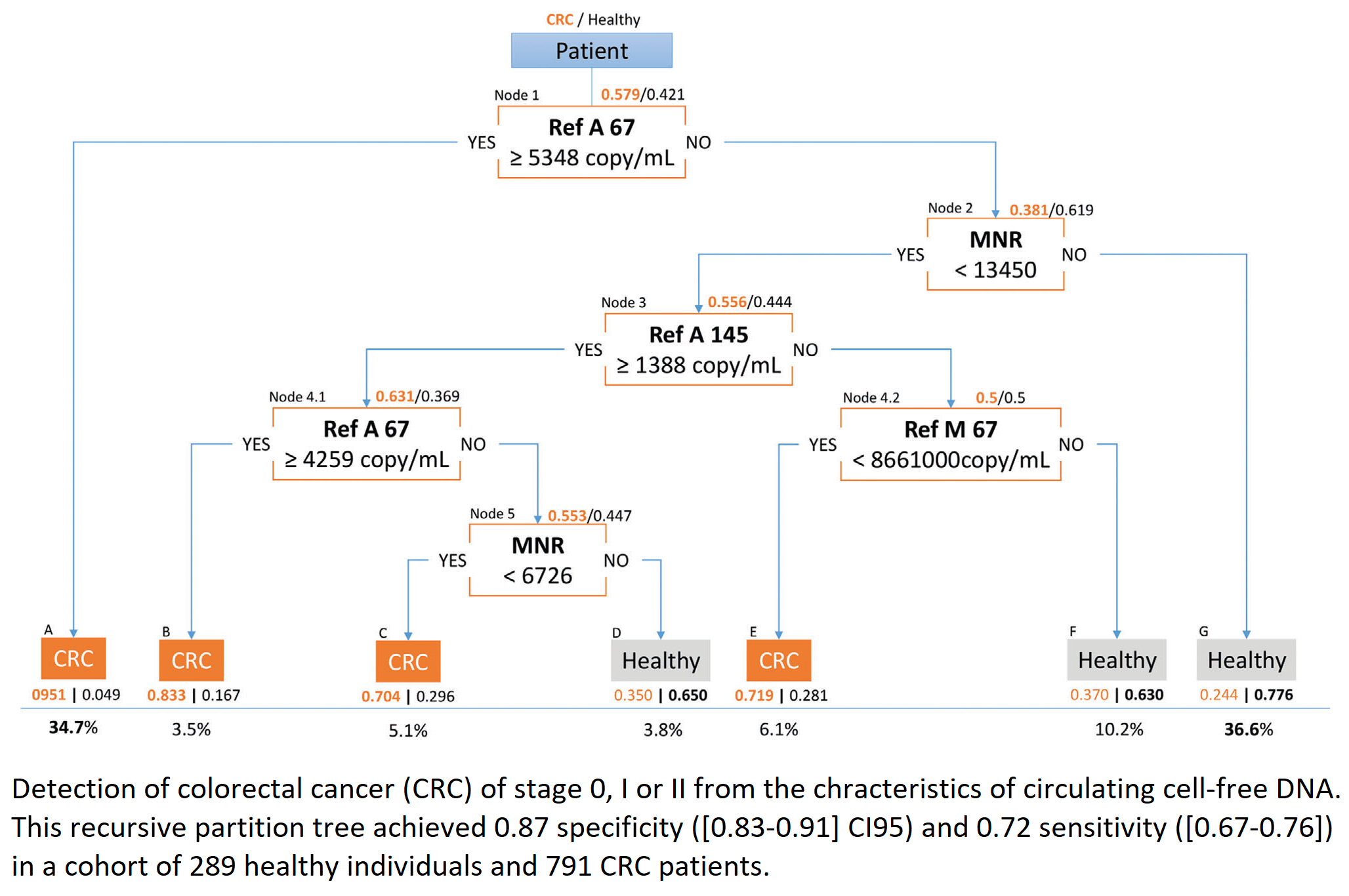
Un autre exemple concerne l'ADN circulant dans la détection du cancer (Tanos, Adv Sci, 2020). Nous avons aussi appliqué des notions de statistiques spatiales et de ML pour étudier la relation entre l'hétérogénéité spatiale de la rigidité de la tumeur et des paramètres cliniques (Lopez-Crapez, Sci Rep, 2022), ou du ML classique pour caractériser le microenvironnement tumoral (Benhamouda, Clin Cancer Res, 2022).
Axe 3 : Applications de la biologie des systèmes (responsables : Cornillot & Colinge)
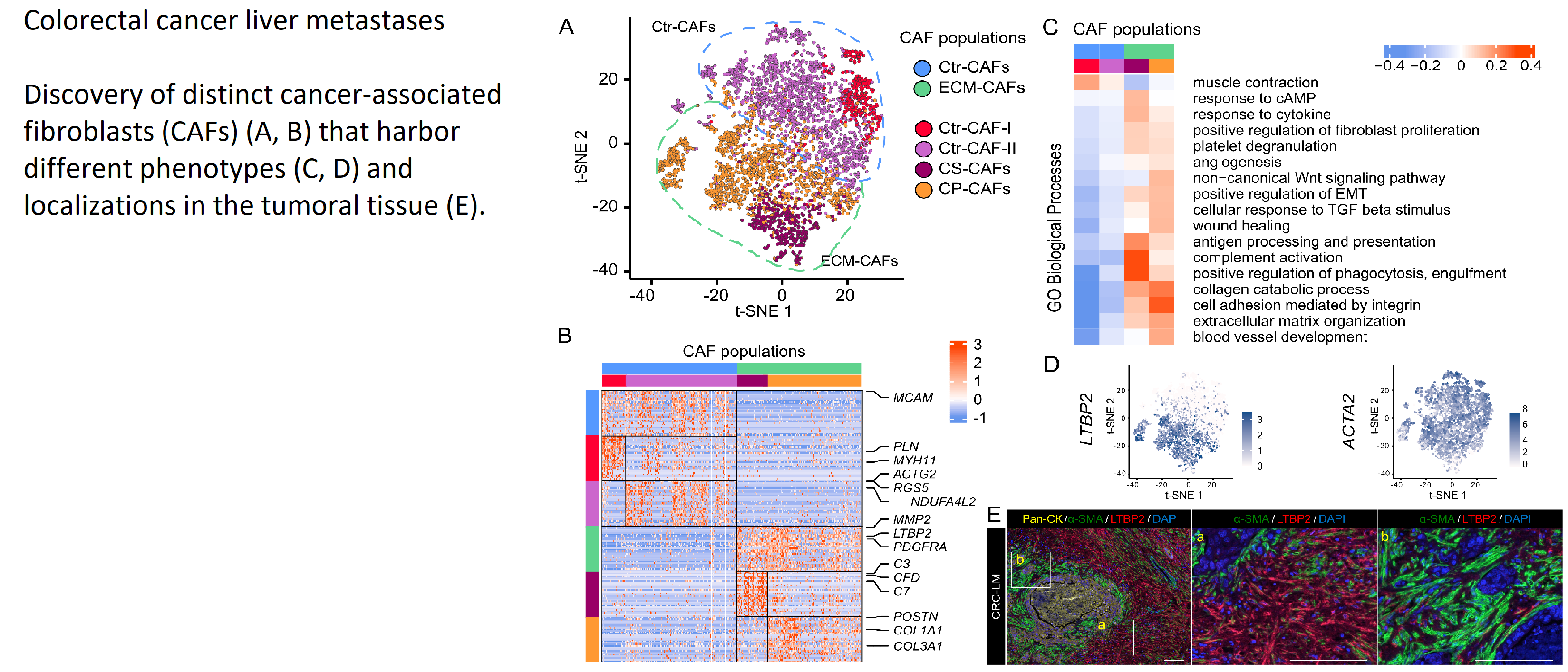
Le développement et l'application de méthodologies computationnelles avancées en génomique fonctionnelle conduit à maintes collaborations avec des biologistes et cliniciens. Les études typiques de cette catégorie consistent en des cohortes de tumeurs de petite à moyenne taille ou des modèles biologiques spécifiques (Alame, Theranostics, 2020 & 2021 ; Giguelay, Theranostics, 2022 ; Souche, Endoscopy, 2021 ; Honda, Theranostics, 2024 ; Figarol, Nat Comm, 2024 ; Singh, Nat Microbiol, 2023 ; Rabia, Front Immunol, 2023). Nous combinons généralement l'intégration de données avec des méthodes des Axes 1 et 2 ou des outils existants par ailleurs.


Situé dans le Parc Euromédecine, l'Institut de Recherche en Cancérologie de Montpellier (U1194) est localisé sur le Campus Val d'Aurelle (ICM, Institut régional du Cancer Montpellier / Val d'Aurelle).
Centre de Recherche U1194 soutenu par l'Inserm, l'Université de Montpellier et l'Institut du Cancer de Montpellier (ICM)
 Institut de Recherche en Cancérologie de
Institut de Recherche en Cancérologie de


